摘要:本文綜合闡述無機高分子絮凝劑與傳統混凝劑在化學特征和凝聚—絮凝行為機理上的差異。PAC的主要成分聚十三鋁在預制條件下才能大量生成,它們對水解反應有一定的穩定性,直接吸附在顆粒物表面,發揮強烈的電中和及粘結架橋作用,其流動電流、ζ電位、絮凝指數及凝聚—絮凝區域圖等,均與傳統混凝劑有很大不同,其計算模式應根據表面絡合及表面沉淀原理建立。
聚合氯化鋁(PAC)作為新型水處理劑,自六十年代以來,頗有取代傳統的硫酸鋁之趨勢,但其絮凝效能顯著高于硫酸鋁的根本原因并未清楚闡明,更無定論,這在一定程度上影響其生產和應用向更高階段發展。
一種通常的推論認為,傳統藥劑直接投放于水中,其水解生成形態由于受到水質等條件的制約,不能形成較有效的絮凝形態和發揮其較高效能,而聚合氯化鋁在人工控制的條件下預制生產,可以達到預期的較佳狀態,投放入水即可發揮電中和及架橋的優異絮凝作用。這種設想雖然有合理性,但并不一定完善,而且未得到實驗證實,尚有很大研究討論余地。
在長期的研究工作中,我們發現人工預制的鋁或鐵聚合物,從形態、特性、功能到作用機理,都與傳統的鋁鹽或鐵鹽混凝劑有很大不同,應看作是新一代制品,需要建立自己的基礎理論和技術工藝體系。本文選取聚合鋁的若干實驗實例來闡述這一問題。
1、鋁水解中間產物的化學形態
鋁鹽在溶液中進行水解→絡合→聚合→膠凝→沉淀→晶化這一系列轉化過程時的化學形態是歷年大量文獻的研究內容,特別是對其水解→絡合→聚合的溶解態中間產物形態爭論甚多而難以統一,這主要表現在兩類觀點上:
(1)鋁化合態的傳統研究方法是化學分析法和電位滴定法。提出水解形態的連續變化分布系列,其羥基化合態由單體到聚合體,按六元環的模式發展,直到生成沉淀仍保持著拜耳石的結構。這種觀點的理論基礎是多核絡合物的核鏈(core-links)絡合機理,在數十年中占統治地位,但尚缺乏直接的結構鑒定證明。
(2)近年來興起的核磁共振AlNMR法和小角度X-射線衍射法的鑒定結果則不同于上述觀點,提出只存在單體、二聚體、和Al13即Al12AlO4(OH)247+及更高聚集體的形態分布模式。這類測定結果仍存在一些未知部分,雖Al13的存在已得到更多的承認,但尚不能說明溶液中形態生成和轉化的完整過程。
即使目前,各研究者的爭論并未取得一致,這主要因為他們的實驗研究結果是在不同條件下采用不同儀器鑒定取得的,特別是他們的樣品是在不同物理化學環境中以不同操作制備的。對形態如此多變的化合態,如果不用綜合分析方法很難得到共同認識。
現在尚沒有一種能夠確切測定Al(Ⅲ)聚合物形態的鑒定方法,其中比較簡單實用的是Ferron逐時絡合分光光度法。Ferron試劑與Al(Ⅲ)的單體及不同聚合物有不同的反應速率。從而把它們分為三類,即:(1)Ala包括單體及初聚物(Al1-3);(2)Alb包括低聚物(如Al6-8)和中聚物(Al13);(3)Alc包括高聚物(Al>13)和溶膠態[nAl(OH)30],這種方法雖只粗略分類,并不能判斷上述兩類觀點,但卻能測定Alb的含量,有很大的實用價值。
表1 Ferron法和Al NMR法測定結果比較
| AlT(M) | B | Ferron/% | NMR/% | Al13/Alb | ||||
| Ala | Alb | Alc | Alm | Al13 | Alu | |||
| 0.100 | 1.0 | 66.83 | 32.68 | 0.49 | 61.43 | 32.88 | 5.69 | 1.01 |
| 0.125 | 1.5 | 48.92 | 50.42 | 0.66 | 39.28 | 51.18 | 9.54 | 1.01 |
| 0.111 | 2.0 | 22.81 | 73.39 | 3.80 | 16.46 | 74.76 | 8.78 | 1.02 |
| 0.100 | 2.5 | 12.48 | 82.85 | 4.67 | 9.75 | 82.92 | 7.33 | 1.00 |
注:AlT=總量,Alm=單體+二聚體,Alu=NMR未測定部分。
表1是我們的實驗結果,表明Ferron法測定的Alb%與核磁共振(NMR)法測定的Al13%十分相近,Al13/Alb≈1。由此可見,在一定條件下,可以認為Alb=Al13,以Ferron法測定Alb即可判定Al13相對含量。不過,在不同的制備條件下,Alb中仍可能含有Al13以外的其他形態。
2、聚十三鋁的生成機理
一般認為聚十三鋁(Al13)是聚合鋁中的較佳凝聚—絮凝成分,其含量可以反映制品的有效性,因而高含量Al13成為聚合鋁制造工藝追求的目標。
在實驗室和水處理工藝中,傳統混凝劑的應用通常是把鋁鹽直接溶于水中或把鋁的濃溶液再加以稀釋。聚合氯化鋁的制備方法很多,常是向鋁溶液中加入強堿液或固體堿,或是在不足量強酸中溶解氫氧化鋁凝膠、金屬鋁、鋁礦粉等。
近年來對Al13的生成機理研究又有新的重要進展,有人提出其生成過程需要有Al(OH)4-作為前驅物。在Al12AlO4(OH)247+ 的核環(Keggin)結構中Al的四面體構成核心,其外圍是12個八面體,來自溶液中的單體或二聚體,如圖1所示,有四面體結構的Al(OH)4- 離子據認為是在堿的加入點生成的,在加入的強堿與酸性鋁溶液的界面上將有pH值的局部區域突變升高,有可能產生Al(OH)4-并隨后生成聚十三鋁。由此認為Al(OH)4-的存在是生成Al13的前提條件,而此條件在高效聚合氯化鋁預先制備時得到很好的滿足。但是,在鋁鹽直接投入水中溶解時,若pH值較低而Al(OH)4-存在很少,則Al13的生成就有很大限制。

這樣,Al(Ⅲ)在溶液中的形態就有兩種轉化途徑,一種是鋁鹽在水中的自然溶解及水解的過程可以稱為“自發水解”過程,另一種則是鋁鹽溶液中加入強堿形成局部pH值升高而強烈水解的過程可以稱為“強制水解”過程。圖2中的(A)和(B)分別表示這兩種不同形態轉化途徑。
表2不同方法制備的溶液中Ala,Alb,Alc的分布
| 制備方法 | 濃度/mol·l-1 | B | Ala/% | Alb/% | Alc/% |
| AlCl3溶液 | 0.100 | 0 | 97.20 | 2.80 | 0 |
| Al2(SO4)3溶液 | 10-4.7 | 70-75 | 5 | 20 | |
| 0.5mol·l-1NaOH以0.04ml·min-1的速度緩慢注入 |
0.100 0.125 0.111 0.100
|
1.0 1.5 2.0 2.5
|
66.83 48.92 22.81 12.48
|
32.68 50.42 73.39 82.85
|
0.49 0.66 3.80 4.67
|
| 堿液一次投入,凝膠在80℃強烈攪拌下溶解 |
0.125 0.125 0.125 0.125
|
1.0 1.5 2.0 2.5
|
58.98 45.38 26.77 15.36
|
36.98 50.40 66.35 55.38
|
4.04 4.22 6.48 29.26
|
| 投入固體碳酸鹽粉,在強烈攪拌下溶解 |
0.125 0.125 0.125 0.125
|
1.0 1.5 2.0 2.5
|
88.02 63.04 52.78 16.86
|
10.74 28.20 33.00 20.68
|
1.26 7.96 14.22 62.46
|
許多實驗研究表明,在直接投加鋁鹽的純溶液中,Alb含量很低而且不一定是聚十三鋁,例如表2中AlCl3和Al2(SO4)3的溶液,其主要成分是Ala,Alb甚少且不一定是Al13。慢速注入法可得到較多的Al13,但不應理解為由于反應進行緩慢,而是由于強堿微滴與鋁液間有大量界面而pH發生局部突變,可產生甚多Al(OH)4-的緣故。一次加堿和固體粉末加堿時生成的Alb都比慢速注入要少,在大規模生產條件下如何生成更多的Al(OH)4-以及Al13應是工藝發展的方向。
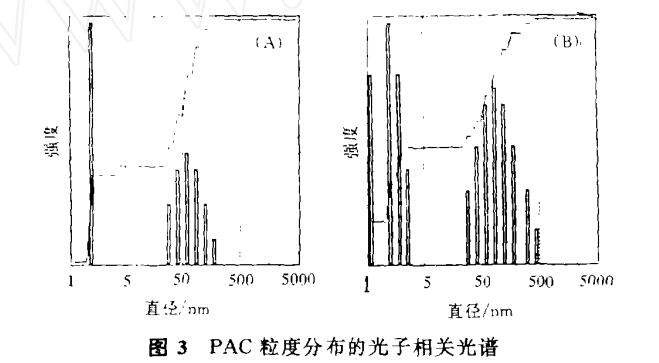
單個Al13的粒度已鑒定為約2.5nm,它們時常結合成線型及枝狀的聚集體,其異形系數約為1-2。圖3是以光子相關法(PCS)得到的激光光散射圖譜,它反映了兩種PAC的粒度分布。圖3A為本實驗室制備的PAC,B=2.0,[Al]=0.3mol·l-1.一部分單顆粒粒度為2.5nm,聚集體的尺寸在40-300nm范圍。圖3B是唐山工業產品,B=1.8,溶液為[Al]=0.3mol·l-1。與圖3A大致相似,但存在一部分小聚集體在3-4nm,大部分聚集體在40-500nm,1nm附近可能是低聚物的響應值。
3、Al(Ⅲ)化合態在水中的穩定性
鋁鹽混凝劑在應用時投入水中后,將被混合稀釋到10-4-10-5mol·l-1,pH升高到6-7以上。根據反應動力學研究,水解和吸附均在微秒級發生,聚合物在1s后生成,Al(OH)3(am)在1-7s中生成。各反應步驟的層次以及與顆粒物的電中和作用將決定于藥劑品種、水的化學組成及顆粒濃度、攪拌的方式與強度等等。
傳統混凝劑在濃溶液中的化合態主要是Ala即單體和初聚體,很少Alb聚合物。在投加入水后,由于稀釋及pH值升高,將迅速發生水解,向生成初聚體及低聚體方向進展,大致是按六元環的結構成長,生成低聚物,或者直接轉化生成沉淀物Al(OH)3(am)以及[nAl(OH)3(am)]。在常見水質情況下,生成Al(OH)4-進而生成Al13的機會很少。基本上是按圖2中(A)的途徑發展。
按照圖2中(B)的途徑良好預制的PAC中含有多量的Al13聚合體,它們對水解有較高的穩定性,在投入水中后相當時間內和不同pH的環境中,可以保持其形態不變。

圖4是PAC樣品投入水中后的Ferron逐時絡合比色曲線(2×10-4mol·l-1Al)。在15min的時間內和pH=5.2-9.5的范圍內其化合態的分布很少變化。這說明Al13具有對水解反應的相對穩定性,在與顆粒物相互作用中可體現其原有的較佳形態。

圖5表示PAC(B=2.5)與AlCl3在恒定pH值溶液中以流動電流檢測器測定的結果對比。Al(Ⅲ)化合態吸附在檢測器的運動活塞表面而產生流動電流。兩種藥劑的流動電流都隨投入劑量增多而增加,但PAC總是比AlCl3要大得多,PAC可以更快地響應并迅速轉為正值, 而AlCl3則一直沒有達到零點。流動電流值與ζ電位值呈線性關系,這表明PAC的吸附與電荷狀態均與AlCl3有很大區別。
4、PAC與顆粒物的相互作用
在水處理絮凝過程中,投入的藥劑與水中污染顆粒物相互作用。鋁的各種化合態吸附在顆粒物表面上發生電中和及粘結架橋效應。使微細顆粒能夠聚集而易于從水中分離。傳統混凝劑進入水中后迅速發生水解,生成低聚物以至沉淀物。與此同時,將發生它們在顆粒物表面的吸附過程。預制的PAC及其Al13投入水中后,Al13及其聚集體將在一定時間內保持其原有形態并立即吸附在顆粒物表面,由于其分子量較大而且整體電荷值較高,因而趨向吸附的力量很強,將會優先結合到顆粒物上。

圖6是PAC和AlCl3在高嶺土懸濁液上Al(Ⅲ)化合態的吸附等溫線,pH=5.0-5.2,表示在單位表面積上鋁化合態的吸附量G(10-6mol·m-2)與溶液中剩余鋁平衡濃度C(10-4mol·1-1)的分布關系,吸附曲線均成Langmuir型,其形式與圖6中PAC的ζ電位曲線是相應一致的。因此,PAC的吸附及電中和效應都要比AlCl3強得多。
5、凝聚-絮凝中的表現
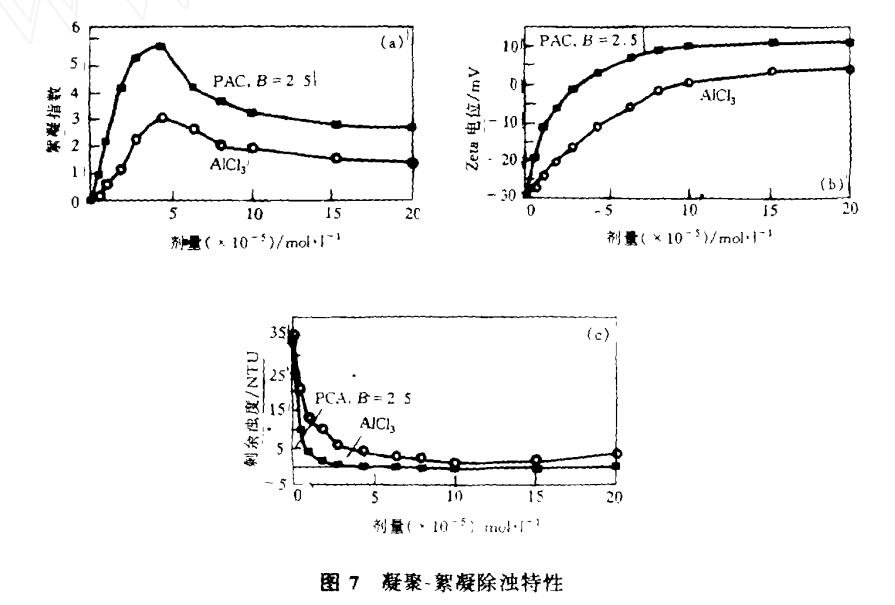
圖7所繪是以PAC和AlCl3在同樣條件下分別去除濁度時,它們的凝聚-絮凝行為和效果的比較。
圖7a是在投藥和混和后生成絮體的絮凝指數(flocculation index)。此參數以光電絮凝檢測儀在線測定,它測定流動狀態下懸濁液的脈動濁度,從而反映聚集后絮體的數目和大小,提供絮凝狀態的信息。測定結果表現為絮凝指數(R):
R=(L/A)1/2(∑NiCi2)1/2
式中,N為數目濃度,C為不同粒度i的絮體散射截面積,L為檢測器光路長度,A為其有效截面積,懸濁液在慢速攪拌后于流動中由檢測器測定,懸濁液pH值為7.3,投藥量為10-5mol Al·l-1。由圖7a可見,在同樣投藥量時,絮體的成長速度受PAC的促進要比AlCl3 快得多。
圖7b為投藥后絮體的ζ電位,樣品在快速攪拌后取出立即用Zetaplus儀測定。
圖7c表示沉淀10min后上澄液的濁度,以光散射濁度儀測定。顯然,以PAC得到的零電荷點及較低濁度所需投藥量都要比AlCl3低得多。
近些年來,許多研究者致力于繪制綜合的凝聚—絮凝區域圖。按照投藥量、pH值及顆粒物表面濃度等參數劃分不同區域,反映和預報凝聚—絮凝狀況和作用機理。我們根據實驗資料,假定剩余濁度降到原水濁度的30%以下即算良好絮凝。繪出相應的凝聚—絮凝區域圖如圖8所示。
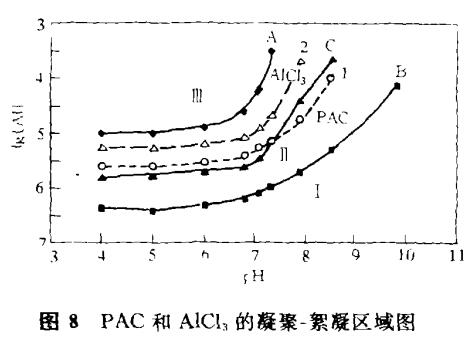
圖8繪出兩種藥劑投加劑量(-1gAlmol·l-1)與溶液pH值相應的良好絮凝區域。曲線A和曲線B之間的區域是PAC的良好絮凝區,曲線1是ZP=0的較優區。另一方面,曲線A與曲線C之間是AlCl3的良好絮凝區,曲線2是其ZP=0的較優區。曲線B的外下方是PAC劑量不足或pH過高的不良絮凝區I。曲線A的上方區域Ⅲ則是投藥過量的再穩定區。顯然,AlCl3的全部良好絮凝區都包括在PAC區內。曲線C的外右下側區域Ⅱ是AlCl3的不良絮凝區,但仍在PAC的良好絮凝區內。
由圖8可見,PAC與AlCl3有不同的絮凝效能和作用機理,預制的PAC有更廣的良好絮凝區,在更低劑量即可達較優除濁效果。
6、無機高分子的絮凝機理
傳統混凝劑的水處理絮凝機理經過歷年大量研究,已趨向于大體一致。一般認為投藥后主要經過水解和吸附過程,絮凝劑生成物發揮電中和及粘結架橋或卷掃作用,其間何種作用為主則與水質及操作條件有關。一般說來,水解反應要比吸附過程更快些。絮凝劑如鋁鹽將以何種化合態吸附在顆粒上,這決定于水質的pH值、顆粒物的濃度及水流擾動狀況等條件,這些作用的歷程都是在微秒及數秒內進行的,實際上是水解反應、吸附過程和流體湍流三種動力學的綜合作用結果。
如果水中顆粒物濃度較高,鋁鹽水解生成的低聚物將成為與顆粒物作用的主要化合態,聚集過程將主要是電中和脫穩型的。如果顆粒物較少而水流擾動強度不夠,與鋁化合態的碰撞接觸將比水解反應緩慢,這時與顆粒物作用的將是鋁的沉淀物或凝膠微粒,聚集過程將主要是粘結架橋以及卷掃沉降型的。這兩種類型是傳統混凝劑的典型狀況,不同水質和物理化學條件下的實際狀況大多是其中間綜合類型。但無論如何傳統混凝劑一般是水解反應先于吸附過程的作用機理,而Al13在一般水質條件下在吸附期間只能少量生成。
預制的PAC及其已生成的Al13投入水中后,Al13及其聚集體將在一定時間內具有穩定性而保持其原有形態,并立即吸附在顆粒物表面,以其較高的電荷及較大的分子量發揮電中和及粘結架橋作用。因此,可以認為高質量的無機高分子絮凝劑在特征上介于傳統絮凝劑與陽離子型有機高分子絮凝劑之間,由于其形態上對水解反應有一定的惰性,其作用機理更接近于有機高分子。但是,這種絮凝劑在本性上仍是多核羥基絡合物的中間產物,相對氫氧化物沉淀是羥基不飽和的。它們與顆粒物的吸附實際是表面絡合配位作用,表面羥基將會適當補充其未飽和位,吸附在表面后,也仍會從溶液中吸取羥基,繼續其水解沉淀過程,直到飽和成為氫氧化物沉淀凝膠,與顆粒物一起生成絮團。因此,無機高分子的凝聚—絮凝機理實際是表面絡合及表面沉淀過程。
吸附的表面絡合理論和計算模式是七十年代以來界面水化學的重要進展, Letter-man、Dentel等用于絮凝過程中,提出傳統絮凝劑的計算模式,他們的作用機理基本概念是:絮凝劑投入水中后,經水解而全部水解生成氫氧化鋁沉淀,實際是帶電荷的氫氧化鋁凝膠吸附在顆粒物表面,發揮凝聚—絮凝作用。無機高分子的作用機理則與此不同,它們是投入水中后主要以Al13直接吸附在顆粒物表面,在表面上繼續水解而轉化為沉淀,由此進行電中和及粘結架橋的凝聚—絮凝作用。因此.無機高分子的絮凝計算模式應是建立在表面絡合及表面沉淀的基礎上,這一作用機理的確證和計算模式的建立將是重要的研究方向。
